分析仪器行业应用解决方案服务商
把握前沿技术,锻造保障能力,追求事业品质
全国服务热线18101957020
本发明属于抗甲状腺癌技术领域,具体涉及一种仑伐替尼杂质及其制备方法和应用。
背景技术:
仑伐替尼(lenvatinib)是由日本卫材公司开发的一种口服多靶点受体酪氨酸激酶(rtks)抑制剂,研究表明,除抑制促血管生成和致癌信号通路相关rtk外,仑伐替尼还能够选择性抑制血管内皮生长因子(vegf)受体的激酶活性,包括vegf1、vegf2、vegf3,以及fgfr、pdgfrα、kit和ret。该药于2015年2月13日获美国fda批准上市,商品名为lenvima,用于治疗局部复发或转移性、放射性碘难治性、进展性的分化型甲状腺癌(dtc)。在欧盟和日本也将仑伐替尼指定用于治疗分化型甲状腺癌。仑伐替尼结构式如下所示:
技术实现要素:
本发明的目的是提供一种仑伐替尼杂质,即式iii化合物。
本发明的另一个目的是提供所述杂质的制备方法。
本发明的最后一个目的是提供所述杂质的应用。
为实现上述目的,本发明提供的技术方案如下:
本发明所述具有式iii所示结构的化合物:
本发明所述具有式iii所示结构的化合物的制备方法,包括:将式ⅱ化合物与3-氯-4-氨基苯酚进行反应,得到式iii化合物,
其中,式ⅱ化合物与3-氯-4-氨基苯酚的摩尔比为1:(0.4~0.5),优选为1:(0.45~0.5),进一步优选为1:(0.48~0.5),更优选为1:0.5。
其中,所述反应在碱性条件下进行,碱是氢氧化钾、氢氧化钠、碳酸钾、碳酸钠、叔丁醇钾、叔丁醇钠、三乙胺、n,n-二异丙基乙胺、吡啶或n,n-二甲基吡啶,优选是氢氧化钾、氢氧化钠、叔丁醇钾或叔丁醇钠。
其中,式ⅱ化合物与碱的摩尔比为1:(1~1.3),优选为1:(1.1~1.2),更优选是1:1.15。
其中,反应溶剂是n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、n-甲基吡咯烷酮、二甲亚砜中的一种或几种。式ii化合物与反应溶剂的比例为1mol:(650~800)ml,优选为1mol:(700~800)ml,进一步优选为1mol:750ml。
其中,反应温度是75~85℃,优选是75~80℃,进一步优选是75℃,反应时间是10~20h,优选12~14h。
其中,所述式ⅱ化合物由式ⅰ化合物与氨水进行氨解反应制备得到:
所述氨解反应的反应溶剂是二甲亚砜、四氢呋喃、乙腈中的一种或几种,式ⅰ化合物和反应溶剂的比例为1g:(2~8)ml,优选1g:(4~6)ml,进一步优选1g:5ml。
式ⅰ化合物和氨水的比例为1g:(10~30)ml,优选为1g:(15~25)ml,进一步优选为1g:20ml。
所述氨解反应的反应温度是100~120℃,优选105~115℃,进一步优选110℃。所述氨解反应的反应时间是5~12h,优选是6~10h,进一步优选是8h。
所述式i化合物由7-甲氧基-4-氧代-1,4-二氢喹啉-6-羧酸甲酯和二甲基亚砜进行反应制备得到。
7-甲氧基-4-氧代-1,4-二氢喹啉-6-羧酸甲酯和二甲基亚砜的比例为1g:(2~6)ml,优选为1g:(3~5)ml,进一步优选为1g:4ml。
所述7-甲氧基-4-氧代-1,4-二氢喹啉-6-羧酸甲酯和二甲基亚砜的反应在催化剂的存在下进行,催化剂是n,n-二甲基甲酰胺或吡啶,7-甲氧基-4-氧代-1,4-二氢喹啉-6-羧酸甲酯和催化剂的比例为1:(0.01~0.05)ml,优选为1g:(0.02~0.04)ml,进一步优选为1g:0.03ml。
所述7-甲氧基-4-氧代-1,4-二氢喹啉-6-羧酸甲酯和二甲基亚砜的反应,反应温度是110~140℃,优选120~130℃,进一步优选125℃。反应时间是2~8h,优选3~5h。
进一步优选地,本发明所述式iii化合物的制备方法,包括以下步骤:
(1)7-甲氧基-4-氧代-1,4-二氢喹啉-6-羧酸甲酯和二甲基亚砜进行反应,得到式i化合物;
(2)式ⅰ化合物与氨水进行氨解反应,得到式ⅱ化合物;
(3)式ⅱ化合物与3-氯-4-氨基苯酚进行反应,得到式iii化合物,
本发明所述的具有式iii所示结构的化合物在制备仑伐替尼原料药或其制剂的杂质对照品中的应用。式ⅲ化合物可作为仑伐替尼的有关物质检测用对照品,用于仑伐替尼及其相关制剂质量控制。
有益效果:1、本发明提供的式ⅲ化合物可以作为杂质对照品控制仑伐替尼原料或制剂的纯度。2、本发明提供的式iii化合物的制备方法操作简单、安全、环保、纯度高。
附图说明
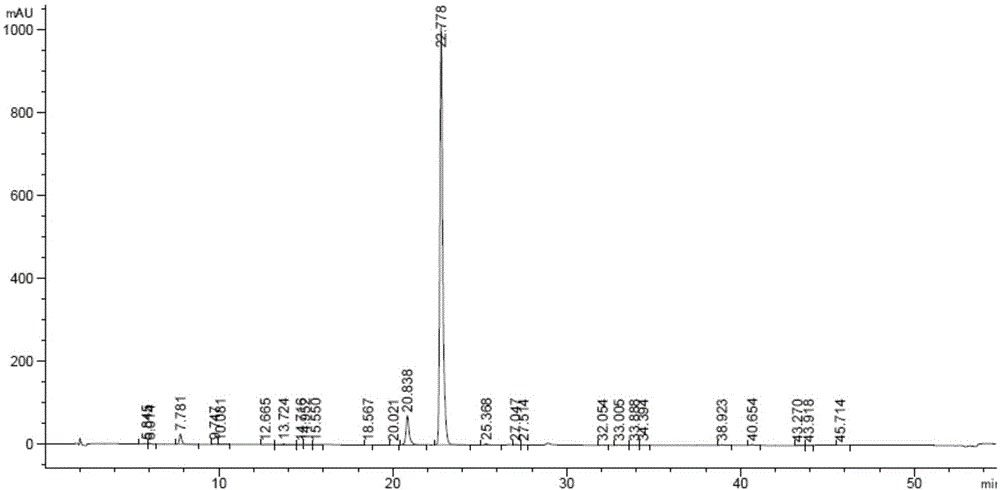
图1是实施例11制备得到的式iv化合物的hplc谱图;
图2是实施例11制备得到的仑伐替尼的hplc谱图。
具体实施方式
本发明将于下文通过实施例更加详细的描述,这些实施例示例性地用于进一步说明,且不应当视为对本发明的限制。
使用bruker的光谱仪在室温记录1h-nmr谱。将氘代二甲亚砜用作溶剂,所述溶剂包括四甲基硅烷作为内标。使用安捷伦6100液质联用仪记录ms谱。使用安捷伦1200hplc进行纯度检查。
实施例1式i化合物的制备
将7-甲氧基-4-氧代-1,4-二氢喹啉-6-羧酸甲酯(5g)在二甲基亚砜(20ml)中溶解,加入0.15mln,n-二甲基甲酰胺,加热至125℃反应3小时,降温至20-25℃,减压浓缩至干。固体溶于二氯甲烷(250ml),倒入饱和碳酸氢钠溶液中(500ml),静置分液,水层用二氯甲烷萃取(250ml)。合并有机层,饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压干燥得4.7g式i化合物。m/z=252[m+h]+。
实施例2式i化合物的制备
将7-甲氧基-4-氧代-1,4-二氢喹啉-6-羧酸甲酯(5g)在二甲基亚砜(20ml)中溶解,加入0.15ml吡啶,加热至125℃反应5小时,降温至25-30℃,减压浓缩至干。固体溶于二氯甲烷(250ml),倒入饱和碳酸氢钠溶液中(500ml),静置分液,水层用二氯甲烷萃取(250ml)。合并有机层,饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压干燥得4.5g式i化合物。m/z=252[m+h]+。
实施例3式ii化合物的制备
将式i化合物(4g)在二甲亚砜(20ml)中溶解,加入氨水(80ml),将反应液加热至110℃,搅拌反应8小时,冷却至常温。过滤,水洗涤(100ml),析出固体减压干燥得3.1g。m/z=237[m+h]+。
实施例4式ii化合物的制备
将式i化合物(4g)在四氢呋喃(20ml)中溶解,加入氨水(80ml),将反应液加热至110℃,搅拌反应8小时,冷却至常温。过滤,水洗涤(100ml),析出固体减压干燥得3.4g。m/z=237[m+h]+。
实施例5式iii化合物的制备
将式ii化合物(5g,0.02mol)和3-氯-4-氨基苯酚(1.5g,0.01mol)在二甲亚砜(15ml)中溶解,加入氢氧化钾(1.3g,0.023mol)。将反应液加热至75℃,搅拌反应12小时。加入水(15ml),冷却至常温并继续搅拌2小时。过滤析出得固体,并用水洗涤(10ml),减压干燥得深紫色固体。所得固体过柱纯化,得棕色固体式iii化合物1.7g,将其进行hplc检测(色谱柱:agilentzobaxbonusrp-c18,3.5μm,4.6*150mm;流速:1.0ml/min;检测波长:243nm;流动相a相:0.05mol/l乙酸铵溶液,b相:甲醇-乙腈(1:1),纯度99.0%。
m/z=544[m+h]+。1hnmr(dmso-d6)δ:9.27(s,1h),8.86(m,1h),8.75(m,1h),8.69(m,1h),8.40(m,1h),7.89-7.55(m,7h),7.41-7.36(m,2h),6.75(m,1h),6.20(m,1h),4.05(s,3h),4.00(s,3h)。
实施例6式iii化合物的制备
将式ii化合物(5g,0.02mol)和3-氯-4-氨基苯酚(1.5g,0.01mol)在n,n-二甲基甲酰胺(15ml)中溶解,加入氢氧化钾(1.3g,0.023mol)。将反应液加热至75℃,搅拌反应12小时。加入水(15ml),冷却至常温并继续搅拌2小时。过滤析出得固体,并用水洗涤(10ml),减压干燥得深紫色固体。所得固体过柱纯化,得棕色固体式iii化合物1.7g,纯度99.2%。m/z=544[m+h]+。
实施例7式iii化合物的制备
将式ii化合物(5g,0.02mol)和3-氯-4-氨基苯酚(1.5g,0.01mol)在n,n-二甲基甲酰胺(15ml)中溶解,加入叔丁醇钾(2.6g,0.023mol)。将反应液加热至75℃,搅拌反应12小时。加入水(15ml),冷却至常温并继续搅拌2小时。过滤析出得固体,并用水洗涤(10ml),减压干燥得深紫色固体。所得固体过柱纯化,得棕色固体式iii化合物1.4g,纯度99.0%。m/z=544[m+h]+。
实施例8式iii化合物的制备
将式ii化合物(5g,0.02mol)和3-氯-4-氨基苯酚(1.5g,0.01mol)在n,n-二甲基乙酰胺(15ml)中溶解,加入叔丁醇钾(2.6g,0.023mol)。将反应液加热至75℃,搅拌反应12小时。加入水(15ml),冷却至常温并继续搅拌2小时。过滤析出得固体,并用水洗涤(10ml),减压干燥得深紫色固体。所得固体过柱纯化,得棕色固体式iii化合物1.4g。纯度99.0%。m/z=544[m+h]+。
实施例9式iii化合物的制备
与实施例5相同,区别仅在于:
式ⅱ化合物与3-氯-4-氨基苯酚的摩尔比为1:0.4。式ⅱ化合物与碱的摩尔比为1:1。式ii化合物与反应溶剂的比例为1mol:650ml。反应温度是80℃。制备得到棕色固体式iii化合物。m/z=544[m+h]+。
实施例10式iii化合物的制备
与实施例5相同,区别仅在于:
式ⅱ化合物与碱的摩尔比为1:1.3。式ii化合物与反应溶剂的比例为1mol:800ml。反应温度是85℃。制备得到棕色固体式iii化合物。m/z=544[m+h]+。
实施例11
仑伐替尼的合成路线如下:
合成路线共3步反应。式ii化合物和3-氯-4-氨基苯酚发生取代反应生成式iv化合物;式iv化合物与氯甲酸苯酯反应生成式v化合物,进一步与环丙胺反应得到式vi化合物仑伐替尼。
申请人研究发现,在仑伐替尼的生产过程,由式ii化合物制备式iv化合物时会产生如式ⅲ所示结构的杂质:
由式ii化合物制备式iv化合物的具体实验步骤如下:
将式ii化合物(10g)、3-氯-4-氨基苯酚(15.8g)和氢氧化钾(6.2g)在二甲基亚砜(60ml)中溶解,氮气保护下加热至60-70℃反应3小时。停止加热,加入水(60ml),搅拌降温至20-25℃,继续搅拌1~2小时。过滤,滤饼用水(50ml)洗涤,减压干燥得14.4g式iv化合物。m/z=344[m+h]+。其hplc谱图见图1,其中,20.838min的出峰对应杂质式ⅲ化合物,22.778min的出峰对应式iv化合物。
式iv化合物的hplc谱图的检测条件为:色谱柱:agilentzobaxbonusrp-c18,3.5μm,4.6*150mm;流速:1.0ml/min;检测波长:243nm;流动相a相:0.05mol/l乙酸铵溶液,b相:甲醇-乙腈(1:1)。
将得到的式iv化合物按照合成路线继续反应,最终制备得到式vi化合物仑伐替尼,制备得到的仑伐替尼的hplc谱图见图2,其中,20.649min的出峰对应杂质式ⅲ化合物,26.414min的出峰对应式vi化合物仑伐替尼。
仑伐替尼的hplc谱图的检测条件为:色谱柱:agilentzobaxbonusrp-c18,3.5μm,4.6*150mm;流速:1.0ml/min;检测波长:243nm;流动相a相:0.05mol/l乙酸铵溶液,b相:甲醇-乙腈(1:1)。
技术特征:
1.一种具有式iii所示结构的化合物:
2.权利要求1所述具有式iii所示结构的化合物的制备方法,其特征在于:将式ⅱ化合物与3-氯-4-氨基苯酚进行反应,得到式iii化合物,
3.根据权利要求2所述的方法,其特征在于:式ⅱ化合物与3-氯-4-氨基苯酚的摩尔比为1:(0.4~0.5),优选为1:(0.45~0.5),进一步优选为1:(0.48~0.5)。
4.根据权利要求2所述的方法,其特征在于:所述反应在碱性条件下进行,碱是氢氧化钾、氢氧化钠、碳酸钾、碳酸钠、叔丁醇钾、叔丁醇钠、三乙胺、n,n-二异丙基乙胺、吡啶或n,n-二甲基吡啶,优选是氢氧化钾、氢氧化钠、叔丁醇钾或叔丁醇钠。
5.根据权利要求4所述的方法,其特征在于:式ⅱ化合物与碱的摩尔比为1:(1~1.3),优选为1:(1.1~1.2)。
6.根据权利要求2所述的方法,其特征在于:反应溶剂是n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、n-甲基吡咯烷酮、二甲亚砜中的一种或几种,式ii化合物与反应溶剂的比例为1mol:(650~800)ml,优选为1mol:(700~800)ml。
7.根据权利要求2所述的方法,其特征在于:反应温度是75~85℃,优选是75~80℃,反应时间是10~20h,优选12~14h。
8.根据权利要求2所述的方法,其特征在于:所述式ⅱ化合物由式ⅰ化合物与氨水进行氨解反应制备得到:
9.根据权利要求8所述的制备方法,其特征在于:所述氨解反应的反应溶剂是二甲亚砜、四氢呋喃、乙腈中的一种或几种,式ⅰ化合物和反应溶剂的比例为1g:(2~8)ml,优选1g:(4~6)ml。
10.根据权利要求8所述的制备方法,其特征在于:所述式i化合物由7-甲氧基-4-氧代-1,4-二氢喹啉-6-羧酸甲酯和二甲基亚砜进行反应制备得到。
11.权利要求1所述的具有式iii所示结构的化合物在制备仑伐替尼原料药或其制剂的杂质对照品中的应用。
技术总结
本发明公开了一种仑伐替尼杂质及其制备方法和应用,所述杂质即为式III化合物。本发明提供的式III化合物可作为仑伐替尼有关物质检测用对照品,用于仑伐替尼原料药或制剂的纯度控制。
技术研发人员:王郑;洪金龙;杨新华;任晋生
受保护的技术使用者:江苏先声药业有限公司
技术研发日:2018.12.27
技术公布日:2020.07.07
热线电话:18101957020
联系人:陈经理
手机:18101957020
邮箱:1436368631@qq.com
地址:上海市浦东新区川沙路1276号2层

联系电话
微信扫一扫